本ガイドは、本剤を適正に使用いただくため、患者の選択、調製方法、蛍光診断法、注意事項等について解説しています。
ご熟読いただき、本剤を適正にご使用いただくためのガイドとしてご活用ください。
2. 禁忌(次の患者には投与しないこと)
| 2.1 |
本剤又はポルフィリンに対し過敏症の既往歴のある患者 |
| 2.2 |
ポルフィリン症の患者[症状を増悪させるおそれがある。] |
| 2.3 |
妊婦又は妊娠している可能性のある女性[9.5、15.2.2参照] |
| 注) |
本資料においては、本剤の含有成分であるアミノレブリン酸塩酸塩(5-ALA HCl)、生物に存在する5-アミノレブリン酸(5-ALA)及び動物実験等に使用した5-アミノレブリン酸リン酸塩(5-ALA P)を総称して「5-ALA」と記載しています。
|
5-ALAは、悪性神経膠腫の腫瘍摘出術中に腫瘍組織を特異的に可視化する光線力学診断用剤です。腫瘍切除術中の残存腫瘍有無の確認に有用な情報が得られます。なお、本剤を用いた診断では、神経機能に関する情報は得られないことを考慮して、切除範囲の決定の参考としてください。
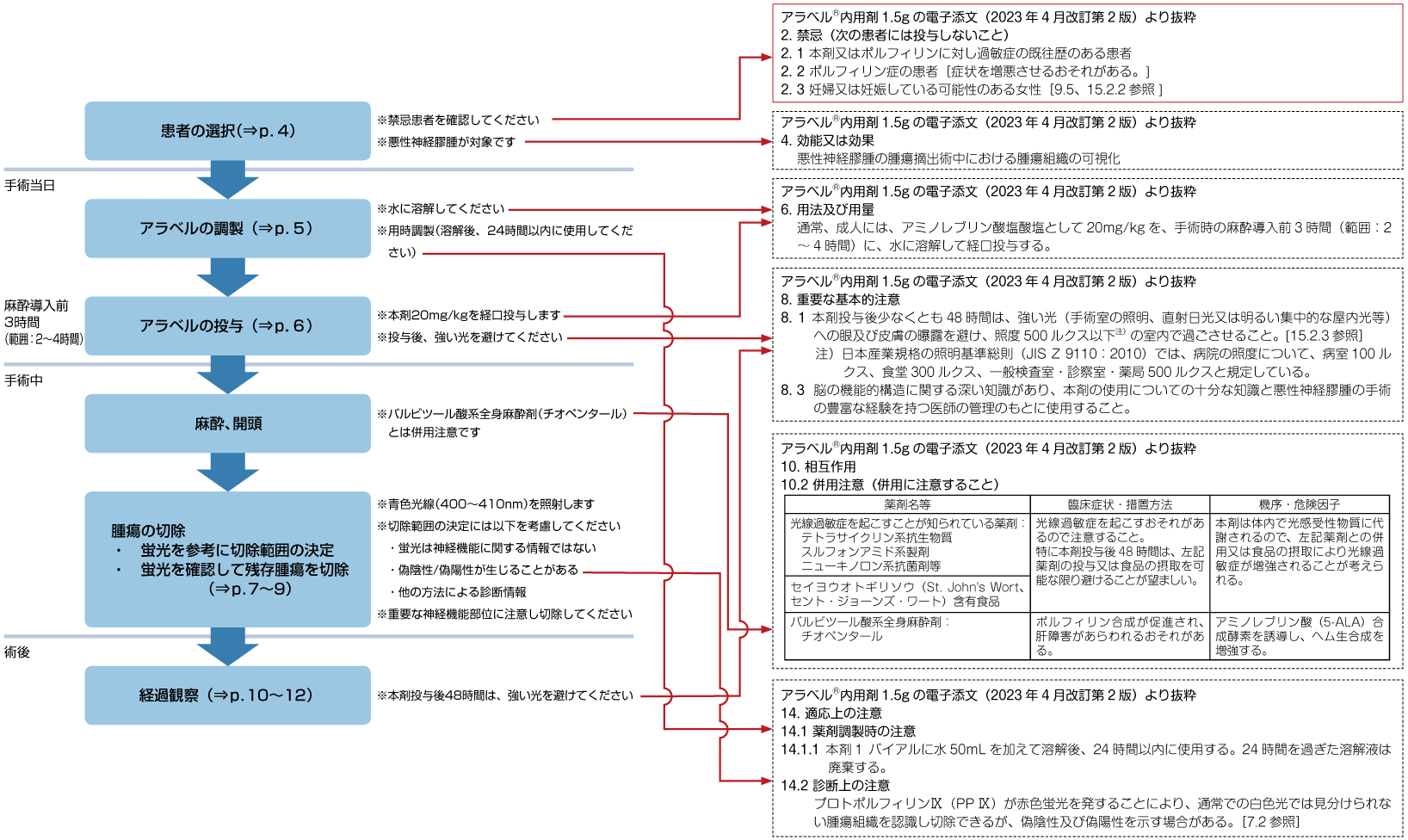
本剤は、脳の機能的構造に関する深い知識があり、本剤についての十分な知識と悪性神経膠腫の手術に豊富な経験を持つ医師の管理のもとに使用してください。[アラベル®内用剤1.5gの電子化された添付文書(以下電子添文)8. 重要な基本的注意 8.3参照]
併せて、最新の電子添文をご熟読ください。
5-ALAは生体内物質であり、生体内ではプロトポルフィリンⅨ(PPⅨ)を経由してヘムが生成されます。
外因性に5-ALAを投与しても同様の経過をたどります。
一方、悪性腫瘍細胞では正常細胞に比べPPⅨが蓄積します。これは、腫瘍細胞では正常細胞に比べてPPⅨを生成する酵素活性が高くPPⅨの生成が促進し、PPⅨからヘムへの生成を触媒する酵素活性が低く、PPⅨの代謝が低下しているためと考えられています。
また、PPⅨは青色光線(400〜410nm)により励起され赤色蛍光(635nm付近)を発する性質を有しています1)。
開頭腫瘍摘出に先立ち患者に5-ALAを経口投与すると、生体内の5-ALAと同様に代謝され、悪性腫瘍細胞ではPPⅨが蓄積されます。術野に青色光線をあてることで、PPⅨが蓄積した悪性腫瘍細胞が赤色蛍光を発するため、腫瘍組織を可視化することができます。
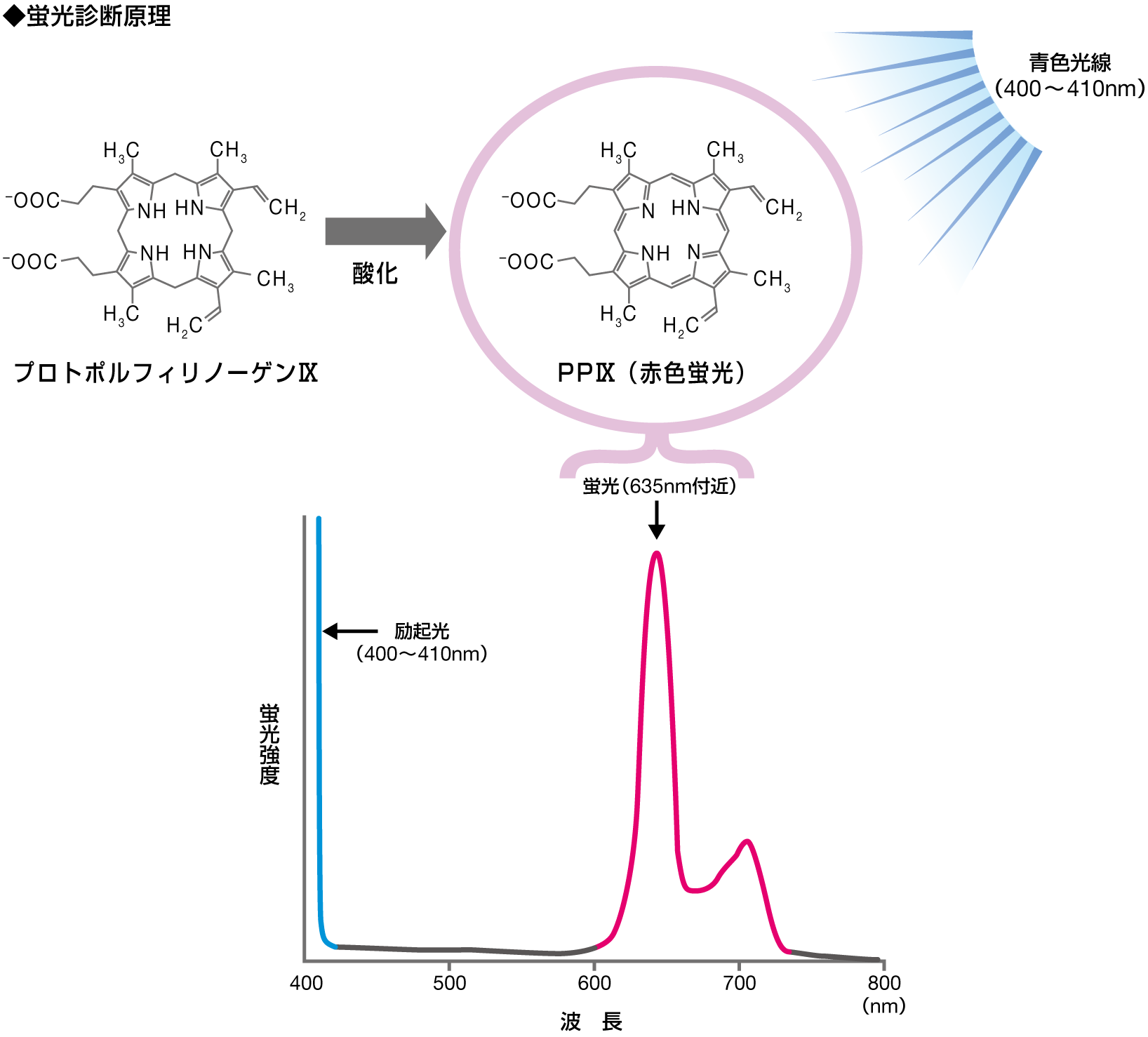
- 1)
- 梶本宜永 他.日本臨牀 2010; 68(S10):375-382. 一部改変
外科的腫瘍切除の適応である悪性神経膠腫患者#
| # |
本剤の有効性を評価した国内第Ⅲ相臨床試験[NPC-07-1]においては、初発及び再発の悪性神経膠腫(WHOグレードⅢ/Ⅳ)の患者を対象として行われました。 |
| 注) |
国内第Ⅲ相臨床試験[NPC-07-1]は、2010年9月〜2011年12月(追跡調査を含む最終検査終了日)に亘って行われ、当時のWHOグレード基準によって治験対象者の適格基準が設定されていました。WHOグレード基準は2021年に改訂され、表記方法もローマ数字から算用数字に変更されています。
|
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
2. 禁忌(次の患者には投与しないこと)
| 2.1 |
本剤又はポルフィリンに対し過敏症の既往歴のある患者 |
| 2.2 |
ポルフィリン症の患者[症状を増悪させるおそれがある。] |
| 2.3 |
妊婦又は妊娠している可能性のある女性[9.5、15.2.2参照] |
注)ポルフィリン :
ビタミンB12やクロロフィル等のポルフィン環を持つ化合物
クロロフィル :
濃い緑色の野菜(ほうれん草、小松菜、ニラ等)に多く含まれる
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
9. 特定の背景を有する患者に関する注意
| 9.1 |
合併症・既往歴等のある患者
|
| 9.1.1 |
心血管系疾患のある患者
収縮期及び拡張期血圧、肺動脈圧並びに肺血管抵抗が低下するおそれがある。
|
| 9.2 |
腎機能障害患者
腎機能障害のある患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
|
| 9.3 |
肝機能障害患者
肝機能障害のある患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
|
| 9.5 |
妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。妊娠ラットに投与した場合、胎児の発育遅延が、また、マウス、ラットの妊娠子宮及び胎児に直接光照射した場合、胎児毒性が生じるとの報告がある。[2.3、15.2.2 参照] |
| 9.6 |
授乳婦
診断上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。
|
| 9.7 |
小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
|
アラベルは凍結乾燥製剤(バイアル入り)です。水に溶解し、経口投与用溶液を調製します。
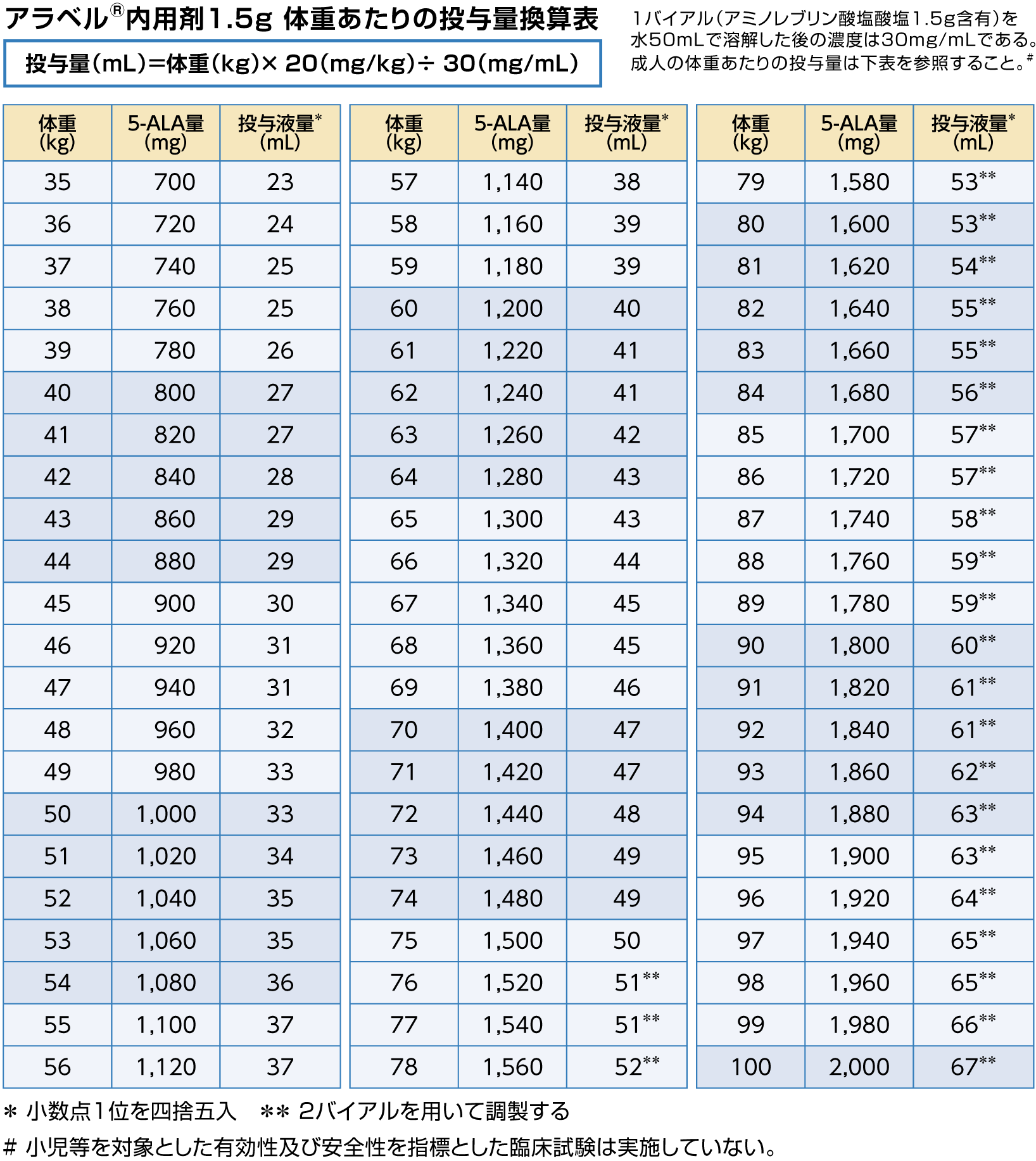
本剤1バイアル(アミノレブリン酸塩酸塩1.5g)に水50mLを加えて溶解後、必要量(20mg/kg)を経口投与してください。
体重が75kgを超える患者には、2バイアルが必要になります。
溶解後は、24時間以内に使用してください。24時間を過ぎた溶解液は廃棄してください。
本剤はバイアル製剤ですが、経口投与する薬剤ですので、注射しないでください。
調製後の溶解液は24時間以内に使用してください。
なお、患者には手術時の麻酔導入前3時間(範囲:2〜4時間)に経口投与させてください。
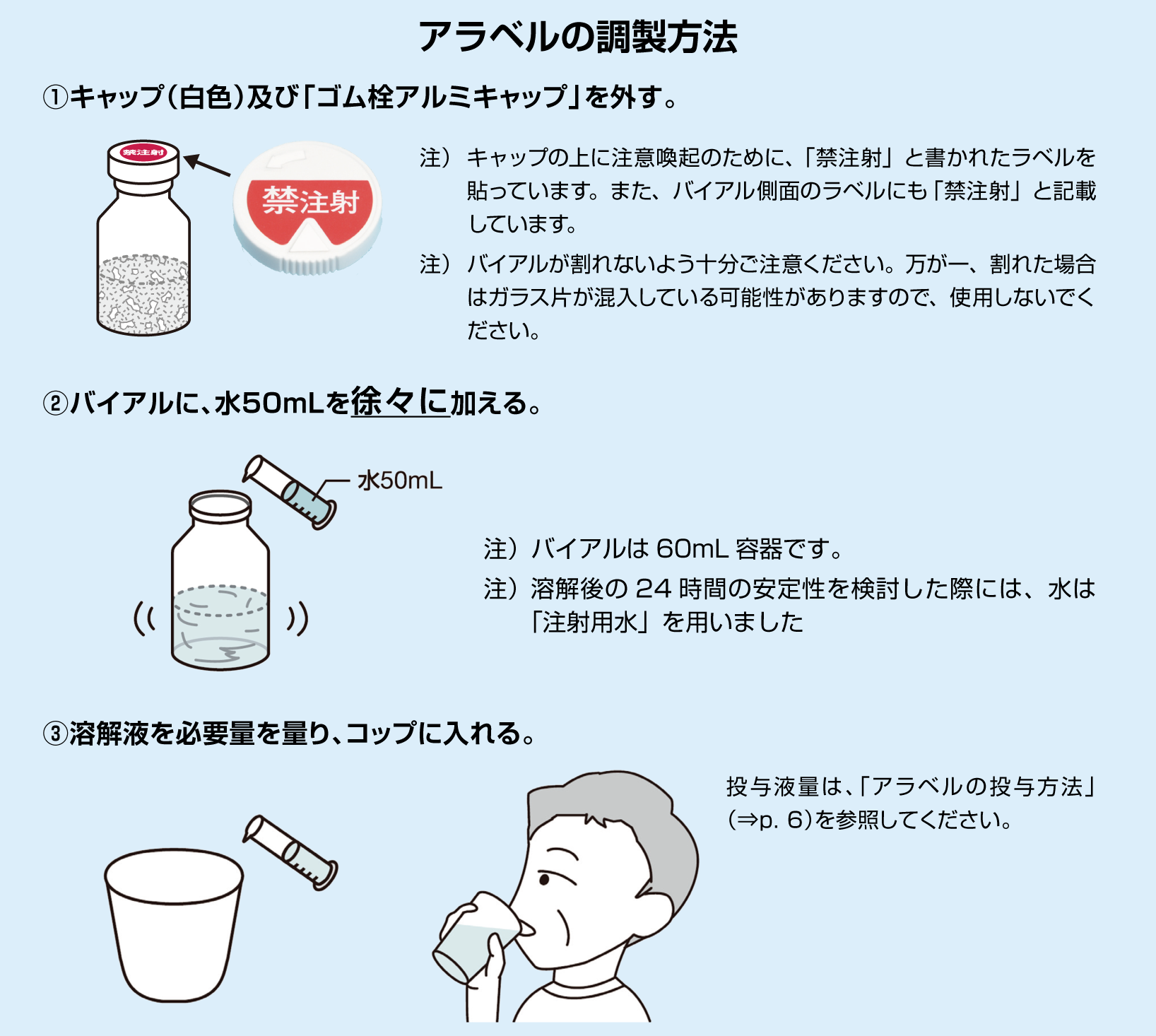
本剤は水に溶解した後、経口投与します。溶解方法は、「アラベルの調製方法」(⇒p.5)を参照してください。
- 通常、成人には、アミノレブリン酸塩酸塩として20mg/kgを、手術時の麻酔導入前3時間(範囲:2〜4時間)に、水に溶解して経口投与します。
- 本剤投与後は、500ルクスを超える光(手術室の照明や直射日光など)に眼及び皮膚が曝露しないよう注意してください。
効能又は効果、用法及び用量、禁忌を含む注意事項等情報等については最新の
DIをご参照ください。
開頭後に青色光線(400〜410nm)を照射することで、腫瘍組織が赤色の蛍光を発し、可視化されます。
光源はキセノンを使用し、高輝度LEDなども使用されることがあります。
蛍光は、その強度により蛍光を強く発する「強蛍光」と弱い蛍光である「弱蛍光」に分けることができます。
国内第Ⅲ相試験2)の結果では、強蛍光領域及び弱蛍光領域の陽性診断率はそれぞれ94.4%(34/36例)及び65.8%(25/38例)でした(臨床データ参照⇒p.15-16)。
(1)
腫瘍摘出方法
国内第Ⅲ相試験2)では、腫瘍切除前に、励起光(青色光線:400〜410nm)を照射することで腫瘍が蛍光を発することを顕微鏡下で確認し、通常の脳腫瘍切除術と同様に白色光下で腫瘍部位を切除しました。その後、再度励起光(青色光線:400〜410nm)を照射して残存腫瘍の有無を確認しました。
(2)
強蛍光及び弱蛍光の判断基準
- 国内第Ⅲ相試験2)における強蛍光/弱蛍光の判断は、術者の主観により行われました。
- 以下の図(写真)◆を判断基準の参考として用いました。
日本レーザー医学会による「脳神経外科疾患を対象としたレーザー治療の安全ガイドライン」では、「(5-ALAを用いた)赤色蛍光を発光する組織の診断は病理学的診断とは基本的に異なるので最終的な組織診断には慎重を要し、病理学的診断を待たなければならない」としています3)。
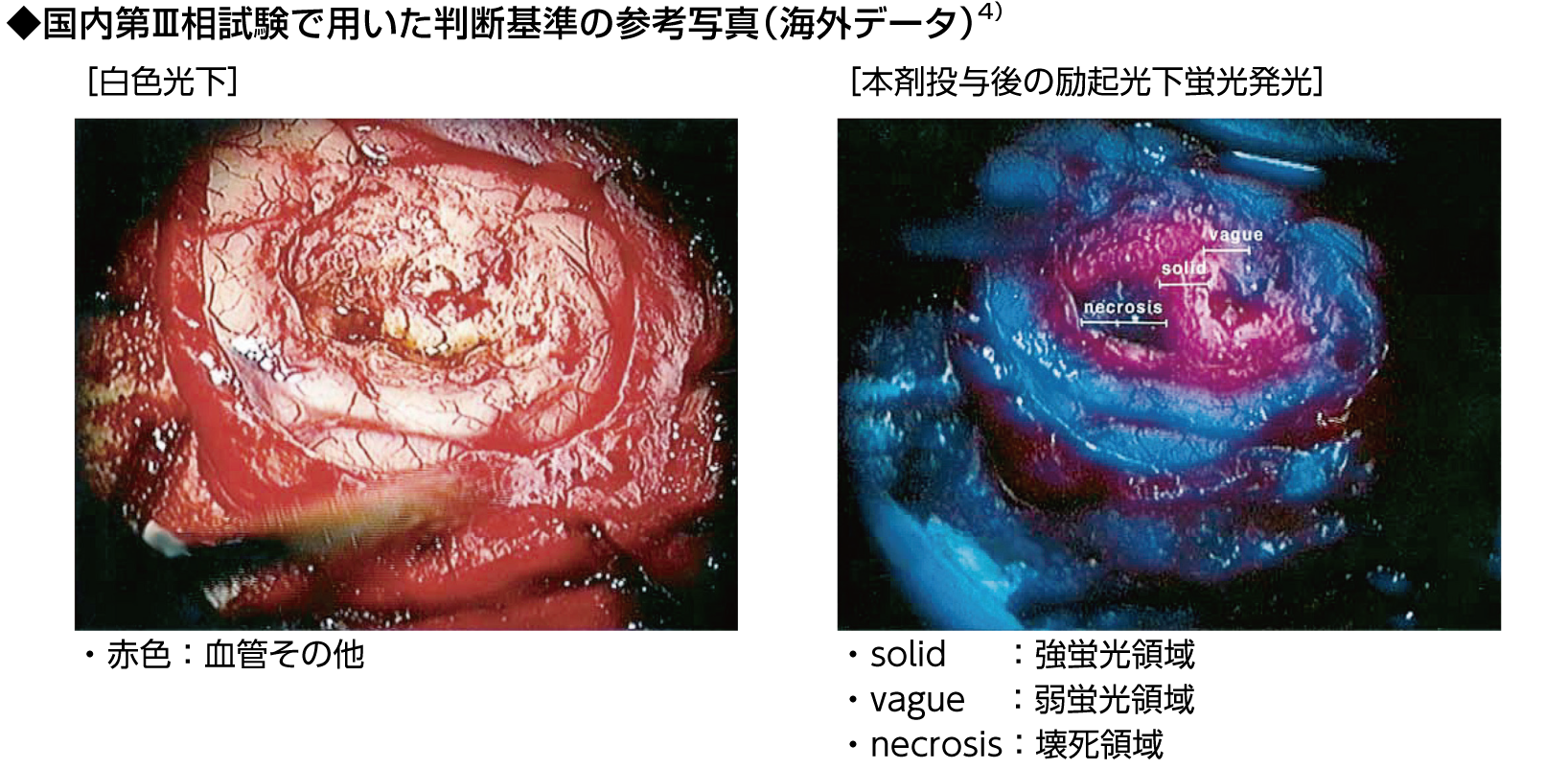
上記は臨床症例の一部を紹介したものであり、全ての症例が同様な結果を示すわけではありません。
- 2)
- 社内資料:国内第Ⅲ相試験(試験番号NPC-07-1)[承認時評価資料]
- 3)
- 脳神経外科疾患を対象としたレーザー治療の安全ガイドライン.日本レーザー医学会誌 2011;S32:44-52.
- 4)
- Stummer W, et al. J Neurosurg. 2000; 93:1003-1013.
(3)
偽陰性及び偽陽性への注意
- 本剤の使用によっても腫瘍組織が可視化されない場合があります。
腫瘍部位に蛍光が認められないことがあります。また、正常組織でも蛍光が認められることがあります。
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
7. 用法及び用量に関連する注意
| 7.2 |
本剤を用いた診断において偽陰性及び偽陽性を示す部位が生じる可能性があることを考慮し、他の方法による診断や残すべき神経機能も踏まえて切除範囲を決定すること。[14.2参照]
|
- 国内第Ⅲ相試験2)では、弱蛍光領域での生検組織ごとの陽性診断率(組織学的に腫瘍細胞が認められた率)は77.2%であり、浸潤している悪性腫瘍細胞と正常細胞が混在していると考えられます(臨床データ参照⇒p.15-16)。
- 本剤投与後、腫瘍部位に蛍光が認められないことがあります。国内第Ⅲ相試験2)45例中3例において、術中迅速病理診断では悪性神経膠腫と判定されたにもかかわらず、本剤投与後腫瘍部位に、強蛍光及び弱蛍光を含めて腫瘍本体の蛍光が認められませんでした。
- 国内第Ⅲ相試験2)では、非蛍光領域には腫瘍が認められないことを確認する目的で、患者に対するリスクが排除される場合のみ採取された蛍光近接領域(非蛍光)及び腫瘍からの遠隔領域(非蛍光)での生検組織ごとの腫瘍細胞ありと判定された割合(陽性率)(患者数38例)を検討した結果、それぞれ44/72検体(61.1%)及び29/61検体(47.5%)でした。この結果から、非蛍光である近接領域及び遠隔領域においても、腫瘍細胞が浸潤していることが示されました(臨床データ参照⇒p.15-16)。
- 2)
- 社内資料:国内第Ⅲ相試験(試験番号NPC-07-1)[承認時評価資料]
(4)
腫瘍切除時の注意点
- 本剤を用いた診断では神経機能に関する情報は得られません。
- 他の方法による診断や残すべき神経機能も踏まえて切除範囲を決定してください。
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
7. 用法及び用量に関連する注意
| 7.1 |
本剤を用いた診断では、神経機能に関する情報は得られないことを考慮して切除範囲の決定の参考とすること。
|
| 7.2 |
本剤を用いた診断において偽陰性及び偽陽性を示す部位が生じる可能性があることを考慮し、他の方法による診断や残すべき神経機能も踏まえて切除範囲を決定すること。[14.2参照]
|
- 本剤によって誘発された脳組織の蛍光発光からは、その組織が有する神経機能に関する情報は得られません。したがって、蛍光を発する組織の切除にあたっては、その領域の神経機能を考慮しながら慎重に判断してください。
- 腫瘍が重要な神経機能の近隣に位置する場合、術前又は術中に、いずれかの測定により腫瘍と機能部位の位置関係の確認を行ってください。
- 本剤投与後48時間は、強い光(手術室の照明、直射日光又は明るい屋内光等)への眼及び皮膚の曝露を避け、照度500ルクス以下の室内で患者を過ごさせてください。
- 肝機能障害があらわれることがあるので、定期的に肝機能検査を行うなど、患者の状態を十分に観察してください。
- 光線過敏症を起こす薬剤の投与やセイヨウオトギリソウ含有食品の摂取は、本剤投与後48時間は可能な限り避けてください。
- 国内第Ⅲ相試験において、安全性を評価した45例中、副作用(臨床検査値異常を含む)発現例数は11例(24.4%)で、悪心3例(6.7%)、嘔吐2例(4.4%)、発熱2例(4.4%)、肝機能異常2例(4.4%)、LDH増加1例(2.2%)、γ-GTP増加1例(2.2%)、リンパ球数減少1例(2.2%)、血小板数減少1例(2.2%)、血尿1例(2.2%)でした。(承認時)
なお、重大な副作用として「肝機能障害」及び「低血圧」があります。
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
8. 重要な基本的注意
| 8.1 |
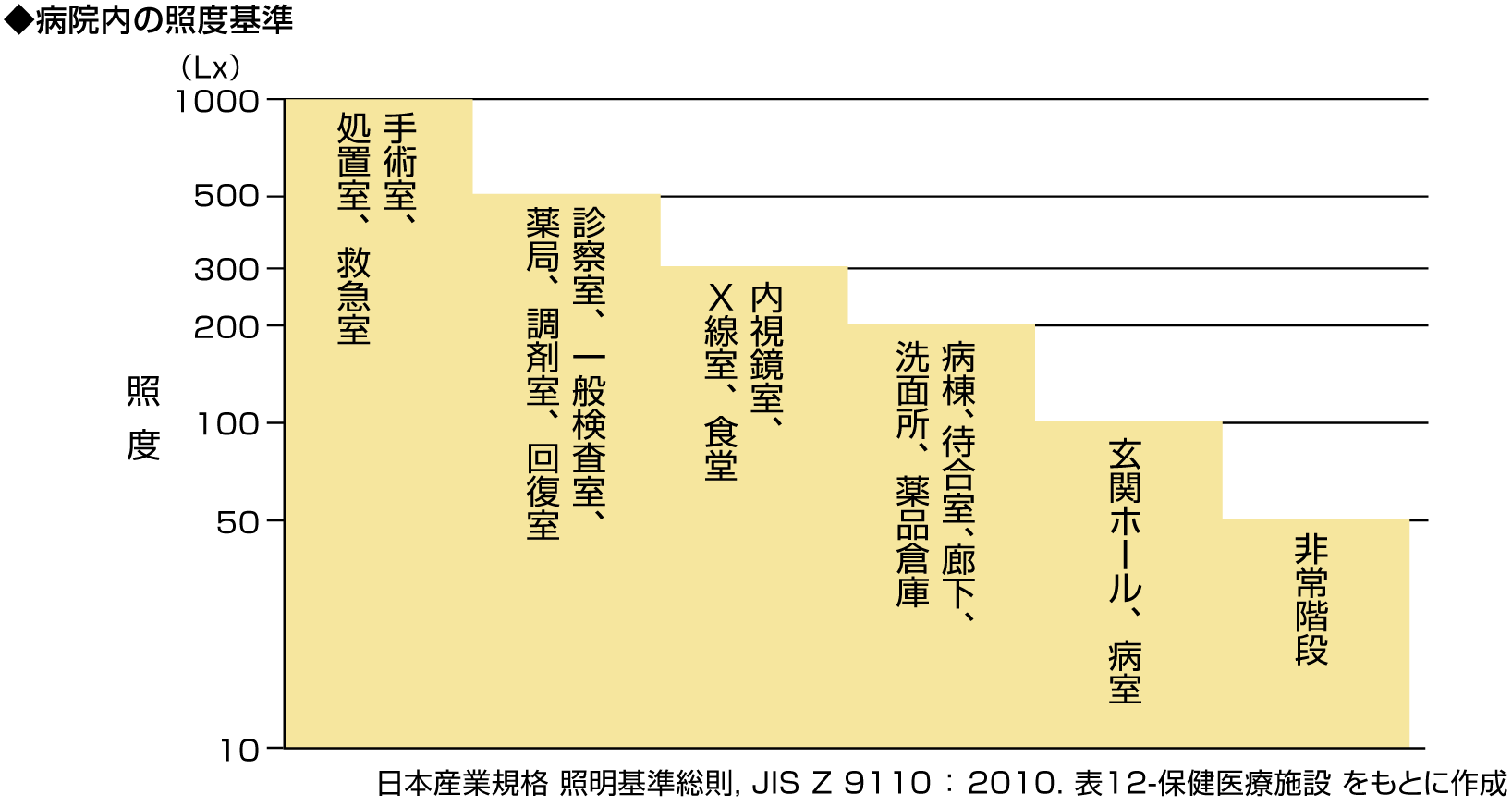
本剤投与後少なくとも48時間は、強い光(手術室の照明、直射日光又は明るい集中的な屋内光等)への眼及び皮膚の曝露を避け、照度500ルクス以下注)の室内で過ごさせること。[15.2.3参照]
| 注) |
日本産業規格の照明基準総則(JIS Z 9110:2010)では、病院の照度について、病室100ルクス、食堂300ルクス、一般検査室・診察室・薬局500ルクスと規定している。
|
|
| 8.3 |
脳の機能的構造に関する深い知識があり、本剤の使用についての十分な知識と悪性神経膠腫の手術の豊富な経験を持つ医師の管理のもとに使用すること。
|
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
10. 相互作用
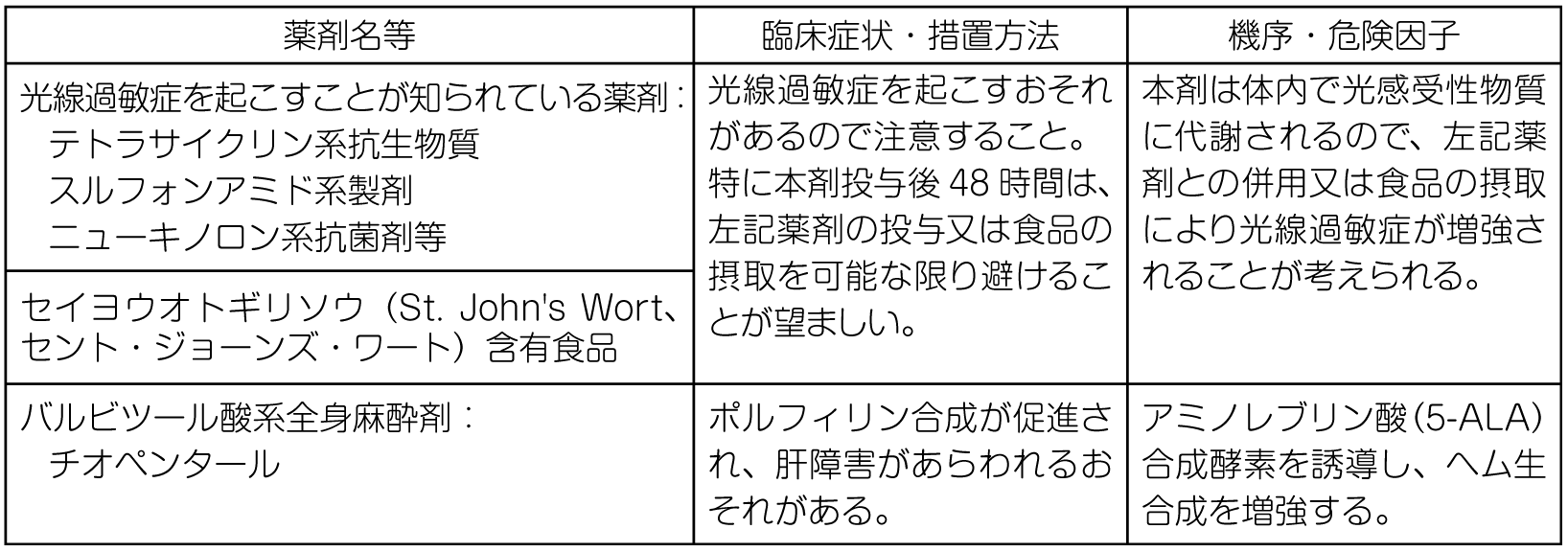
(1)
光への注意
本剤投与後48時間は、照度500ルクス以下の室内で患者を過ごさせてください。
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
8. 重要な基本的注意
| 8.1 |
本剤投与後少なくとも48時間は、強い光(手術室の照明、直射日光又は明るい集中的な屋内光等)への眼及び皮膚の曝露を避け、照度500ルクス以下注)の室内で過ごさせること。[15.2.3参照]
| 注) |
日本産業規格の照明基準総則(JIS Z 9110:2010)では、病院の照度について、病室100ルクス、食堂300ルクス、一般検査室・診察室・薬局500ルクスと規定している。
|
|
15. その他の注意
| 15.2 |
非臨床試験に基づく情報
|
| 15.2.2 |
動物細胞に5-ALAを曝露後、光照射すると遺伝毒性を示すことが報告されている。[2.3、9.5参照]
|
| 15.2.3 |
マウスへの静脈内投与後に紫外線照射すると光毒性(死亡、炎症性皮膚反応)を生ずることが報告されている。[8.1参照]
|
海外の健康成人男性(21症例)を対象とした皮膚感作試験5)では、投与後12、24及び48時間において、背部及び臀部に、8段階の線量の紫外線を照射し、最小紅斑量(皮膚に紅斑を生じる最小の照射熱量)を検討しました。本剤投与後の最小紅斑量は、本剤投与後12時間及び24時間において、投与前に比較し有意に低下していることが認められましたが(p<0.0001,分散分析)、48時間では投与前の値に回復していました。
動物細胞(CHL細胞)に代謝活性化系非存在下において24、48時間連続で5-ALAを曝露後、光照射すると遺伝毒性(染色体の構造異常を有する細胞の出現頻度の増加傾向)が報告されています6)。
マウスへの静脈内投与後に紫外線照射すると光毒性(死亡、炎症性皮膚反応)を生じることが報告されています7)。
- 5)
- 社内資料:海外バイオアベイラビリティ試験(試験番号MC-ALS.20/BV)
- 6)
- 社内資料:ほ乳類培養細胞を用いる染色体異常試験
(2)
肝機能障害
本剤により肝機能障害があらわれることがあるので、定期的に肝機能検査を行うなど、患者の状態を十分に観察してください。
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
8. 重要な基本的注意
| 8.2 |
肝機能障害があらわれることがあるので、定期的に肝機能検査を行うなど、患者の状態を十分に観察すること。[11.1.1、15.2.1参照]。
|
11. 副作用
| 11.1.1 |
肝機能障害(6.7%)
γ-GTP(6.7%)、AST(4.4%)、ALT(4.4%)、Al-P(2.2%)の増加等を伴う肝機能障害があらわれることがある。[8.2、15.2.1参照]
|
15. その他の注意
| 15.2 |
非臨床試験に基づく情報
|
| 15.2.1 |
動物試験(ラット、イヌ)で代謝物(PPⅨ)による肝臓障害が報告されている。[8.2、11.1.1参照]
|
国内第Ⅲ相試験2)で報告された肝機能障害に関連する副作用
-
肝機能異常 2例(4.4%)
異常と判定された検査項目の内訳
1例:
γ-GTP増加、AST増加、ALT増加、AL-P増加
-
γ-GTP増加 1例(2.2%)
動物試験(ラット、イヌ)で代謝物(PPⅨ)による肝臓障害が報告されています8-10)。
- 2)
- 社内資料:国内第Ⅲ相試験(試験番号NPC-07-1)[承認時評価資料]
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行ってください。
(1)
重大な副作用
| 1)肝機能障害: |
肝機能障害があらわれることがあるので、定期的に肝機能検査を行うなど、十分に観察を行い、異常が認められた場合には、適切な処置を行ってください。
|
| 2)低血圧: |
低血圧があらわれることがあるので、十分に観察を行い、異常が認められた場合には、適切な処置を行ってください。なお、手術後も低血圧が遷延することがあるので、十分な観察を継続し、異常が認められた場合には、適切な処置を行ってください。
|
(2)
その他の副作用
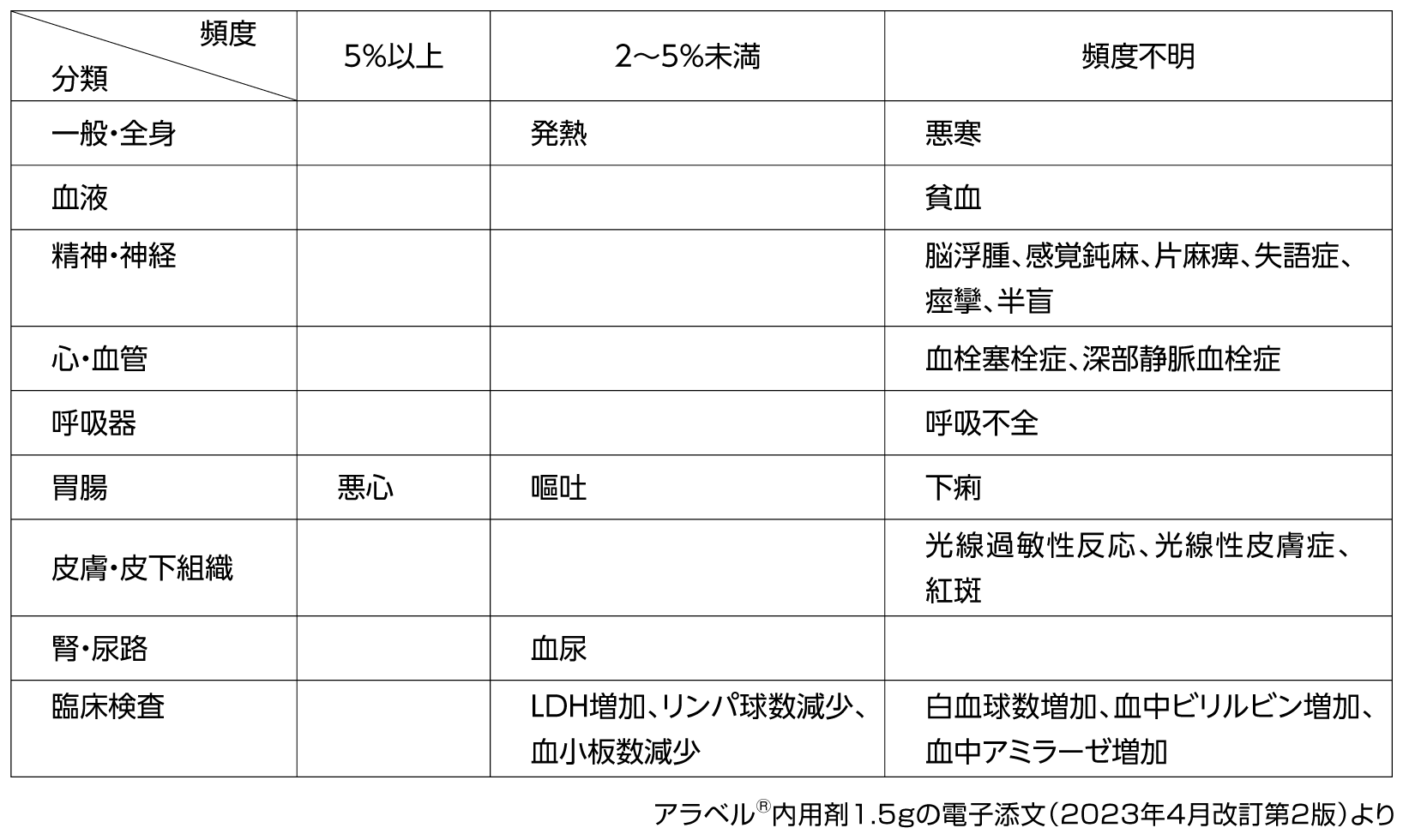
アラベル®内用剤1.5gの電子添文(2023年4月改訂第2版)より抜粋
| 11. |
副作用
|
|
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
|
| 11.1 |
重大な副作用
|
| 11.1.1 |
肝機能障害(6.7%)
γ-GTP(6.7%)、AST(4.4%)、ALT(4.4%)、Al-P(2.2%)の増加等を伴う肝機能障害があらわれることがある。[8.2、15.2.1参照]
|
| 11.1.2 |
低血圧(頻度不明)
手術後も、低血圧が遷延し、昇圧剤の持続投与が必要な症例が報告されている。
|
禁忌を含む注意事項等情報等については最新の
DIをご参照ください。
(1)
試験概要
| 目的 |
初発及び再発の悪性神経膠腫(WHOグレードⅢ/Ⅳ#)を対象として、NPC-07(以下、アラベル)による蛍光切除術の診断能、安全性及び薬物動態の検討を行う。 |
| 試験デザイン |
STEPⅠ及びSTEPⅡの構成による多非盲検・非対照施設共同試験(有効性び病理判定は盲検化)
| STEPⅠ |
少数例で本剤の安全性及び薬物動態を検討する。 |
| STEPⅡ |
症例数を増やし本剤の診断能、安全性を検討する。 |
[STEPⅡへの移行] STEPⅠでの薬物動態及び本剤投与後28日間で発現した有害事象の発現状況等を評価して移行の可否を判断し、7日間以上空けてSTEPⅡに移行した。
|
| 対象 |
放射線学的診断で初発又は再発の悪性神経膠腫(WHOグレードⅢ/Ⅳ#)と推定され、外科的腫瘍切除の適応があり、試験参加への文書同意が得られた18〜70歳の患者45例[解析対象例数:STEPⅠ 10例、STEPⅡ 35例] |
| 投与方法 |
アラベル1.5g(1バイアル)を投与3時間以内に50mLの注射用水で溶解し、悪性神経膠腫手術時の麻酔導入前3時間(範囲:2〜4時間)に、アラベル20mg/kgを単回経口投与した。 |
| 試験期間 |
海外の臨床試験において、5-ALA及びPPⅨの血漿中からの消失は比較的早く、また、有害事象は投与後早期に発現していたが、安全性評価に十分な観察期間が必要と考えられたことから、28日間の観察期間を設定した。 |
| 評価項目 |
| 〈薬物動態(STEPI)〉 |
アラベル投与前、投与後0.5、1、2、3、4、5、6、8、12、24、48時間の血漿中5-ALA及び代謝物であるPPⅨ濃度および薬物動態パラメータ(Cmax、AUCt、tmax 及びt1/2) |
| 〈主要評価項目〉 |
蛍光組織の陽性診断率(蛍光組織の生検組織における腫瘍細胞が全て陽性と判定された患者割合)*、強蛍光及び弱蛍光別の陽性診断率 |
| 〈副次評価項目〉 |
蛍光組織での生検組織ごとの陽性診断率、残存腫瘍のない患者の割合(術後72時間以内のMRI検査による)、蛍光近接領域(非蛍光)及び腫瘍からの遠隔領域(非蛍光)におけるそれぞれの生検組織ごとの陽性診断率、感度と特異度 等 |
| 〈安全性評価〉 |
有害事象、臨床検査値 |
|
| 解析計画 |
顕微鏡下で腫瘍が蛍光を発することを切除前に励起光(青色;光)で確認後、通常の脳腫瘍切除術と同様に白色光下で腫瘍部位を切除した。その後、再度励起光で残存腫瘍の有無を確認した。蛍光が観察されなかった場合は実施しなかった。
アラベル投与全例(45例)を安全性評価対象集団、術中迅速診断で基準(WHOグレードⅢ/Ⅳ#)に合致しない又は腫瘍本体に蛍光がなかった7例を除いた38例を有効性解析対象集団とした。
| 〈主要評価項目〉 |
蛍光組織の陽性診断率及びその95%信頼区間(95% CI)を求めた。蛍光組織の陽性診断率は以下の計算式で求めた。
陽性
診断率
(%) |
= |
全ての蛍光組織が腫瘍細胞陽性
と判定された患者数
蛍光組織検体
採取患者数
|
×100 |
また、強蛍光/弱蛍光別の陽性診断率及びその95% CIを求めた。
|
| 〈副次評価項目〉 |
蛍光組織の陽性診断率は以下の計算式で求めた。
蛍光の質ごとの
生検組織陽性診断率
(%) |
= |
腫瘍細胞数
蛍光した
生検組織検体数
|
×100 |
術後72時間以内のMRI検査により判定した残存腫瘍のない患者の割合は以下の計算式で求めた。
残存腫瘍のない
患者の割合
(%) |
= |
残存腫瘍のない
患者数
全対象患者数
|
×100 |
蛍光近接領域(非蛍光)及び腫瘍からの遠隔領域(非蛍光)におけるそれぞれの生検組織ごとの陽性診断率は、上記の計算方法に準じて求めた。
|
| 〈安全性評価〉 |
有害事象、臨床検査値異常の発現例数、件数、発現率を求めた。 |
|
| # |
本試験は2010年に行われて、当時のWHOグレード基準によって適格基準が設定された。その後、2021年にWHOグレード基準は改訂され、表記方法もローマ数字から算用数字に変更されている。
|
| * |
白色光下で腫瘍部位を切除した後、担当医の主観による強蛍光領域及び弱蛍光領域からそれぞれ最大3箇所の蛍光組織を採取し、最大6検体が全て腫瘍陽性であると判定された患者を陽性診断例として集計した。
|
(2)
薬物動態
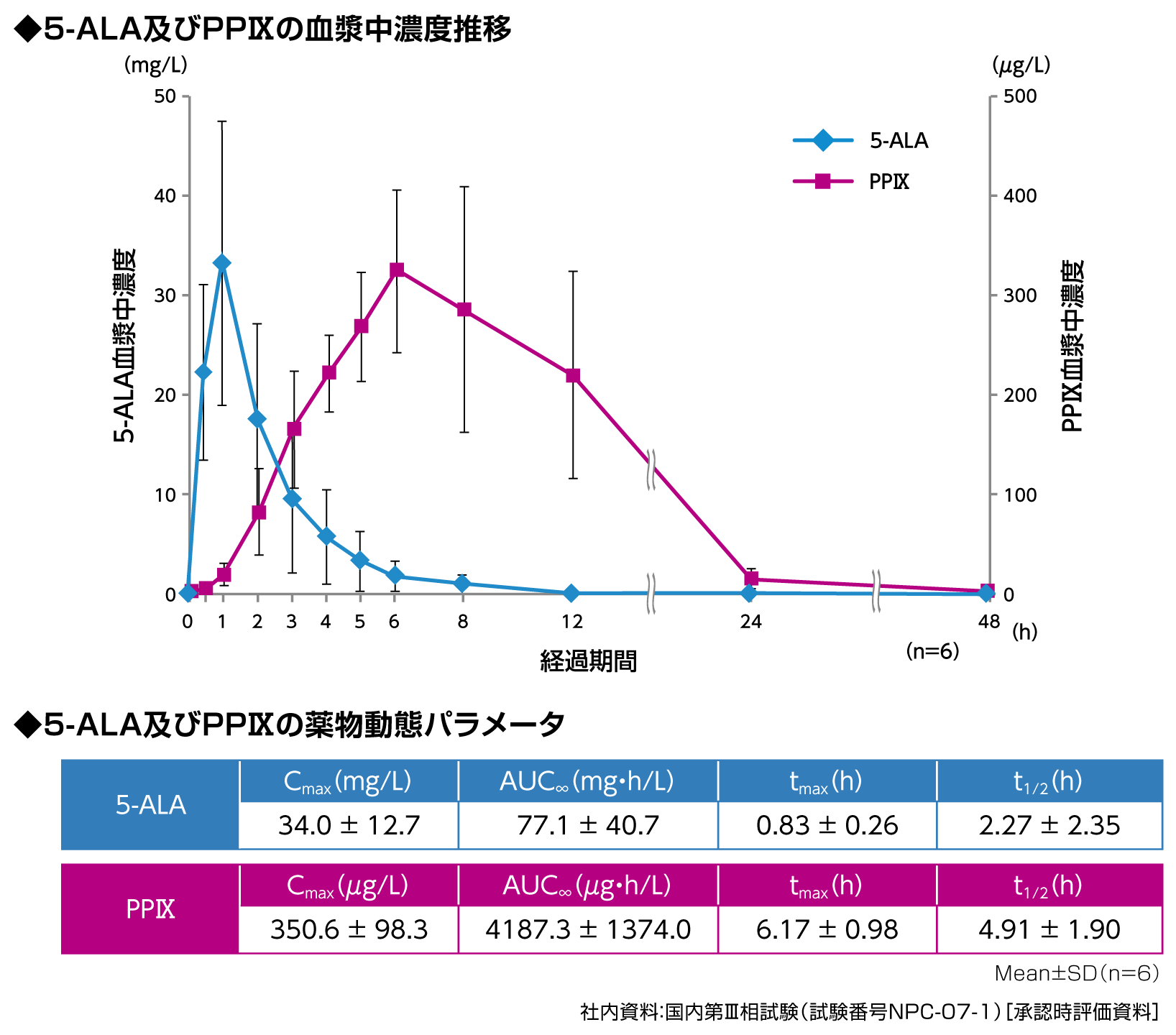
国内第Ⅲ相試験のSTEPⅠにおいて、悪性神経膠腫患者6例に本剤20mg/kgを空腹時単回経口投与したときの未変化体(5-ALA)及び代謝物であるPPⅨの血漿中濃度は下図のとおり推移しました。
5-ALAは、投与後0.83時間に最高濃度34.0mg/Lを示し、投与12時間後にはほぼ投与前の値まで減少しました。消失半減期は2.27時間でした。PPⅨの血漿中濃度は5-ALAに比べ緩やかに上昇し、投与6時間後に最高値に達し、投与48時間後にほぼ投与前の値まで減少しました。
(3)
有効性評価
◆
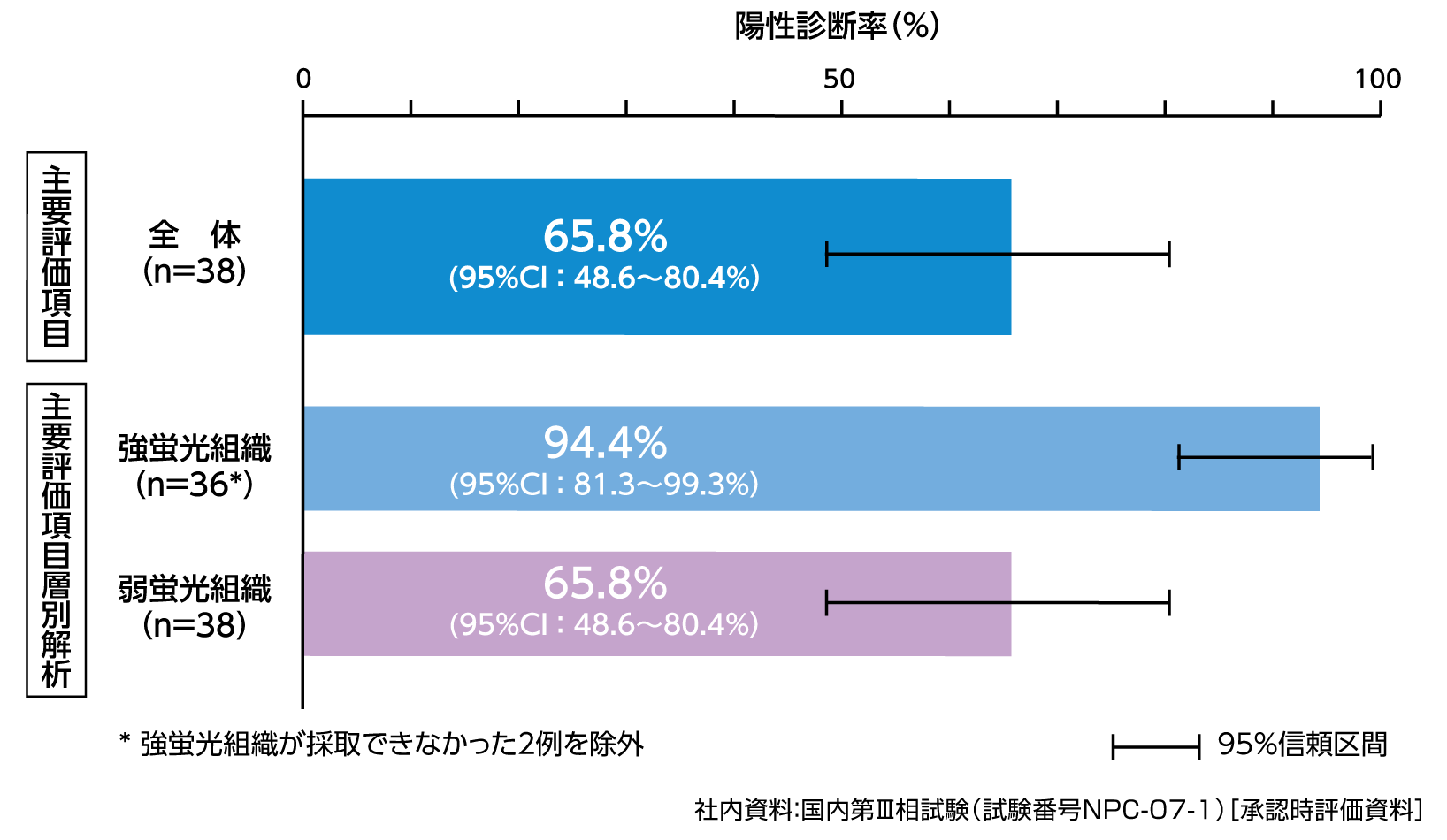
蛍光組織の陽性診断率(患者の割合)(主要評価項目)
有効性解析対象集団38例のうち、蛍光組織の生検標本のすべてが腫瘍細胞と判定された患者の割合(陽性診断率)は65.8%(25/38例、95% CI: 48.6〜80.4%)でした。
また、強蛍光/弱蛍光別の陽性診断率は、強蛍光組織が採取できなかった2例を除く36例における強蛍光での陽性診断率は94.4%(34/36例、95% CI: 81.3〜99.3%)、弱蛍光別の陽性診断率は65.8%(25/38例、95% CI: 48.6〜80.4%)でした。
◆
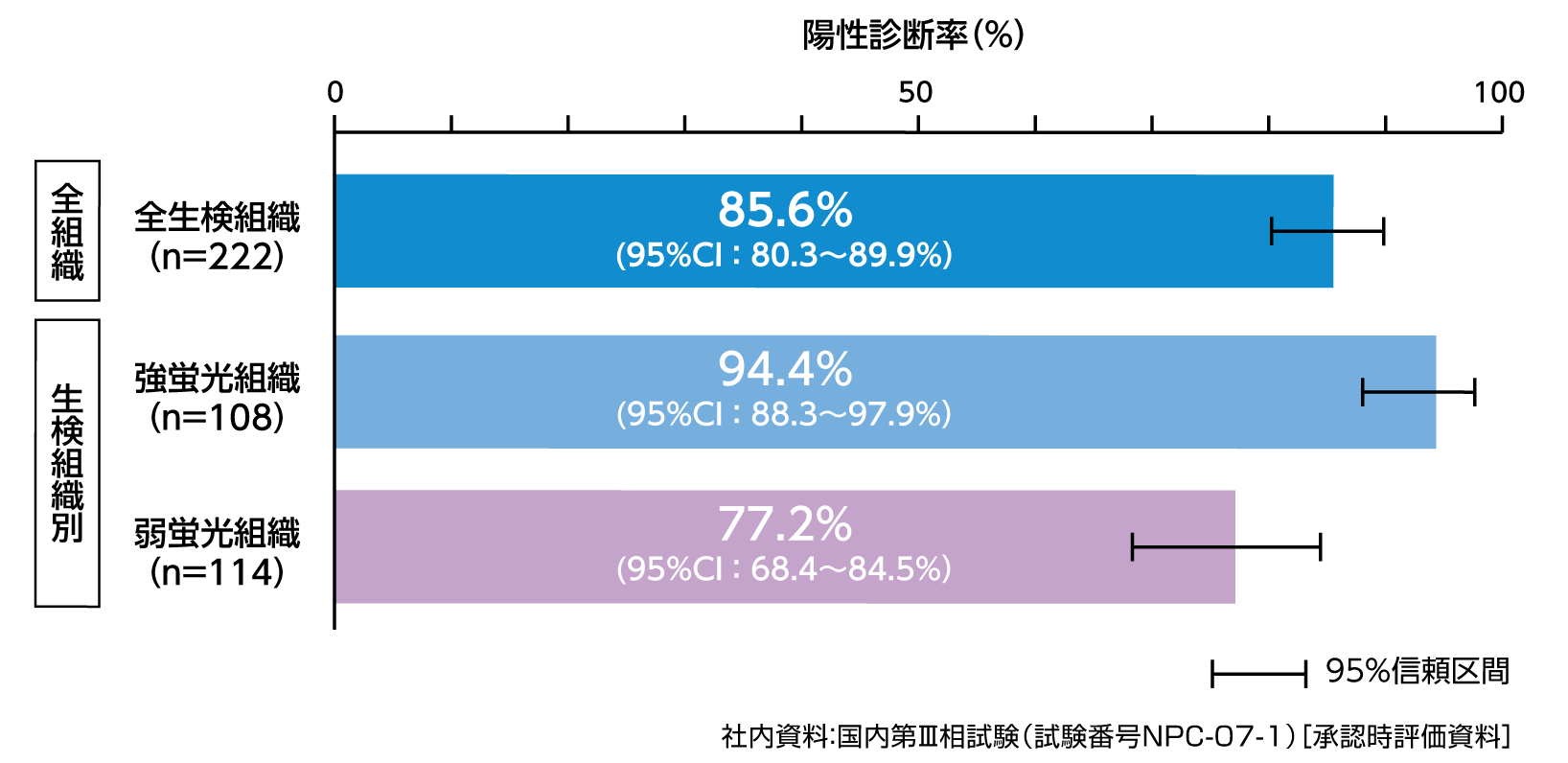
蛍光組織での生検組織ごとの陽性診断率(副次評価項目)
蛍光組織での生検組織ごとの陽性診断率は、強蛍光+弱蛍光の生検組織における陽性率が85.6%(190/222検体、95% CI: 80.3〜89.9%)でした。
強蛍光組織においては、94.4%(102/108検体、95% CI: 88.3〜97.9%)が腫瘍細胞陽性であり、弱蛍光組織では77.2%(88/114検体、95% CI: 68.4〜84.5%)が腫瘍細胞陽性でした。
◆
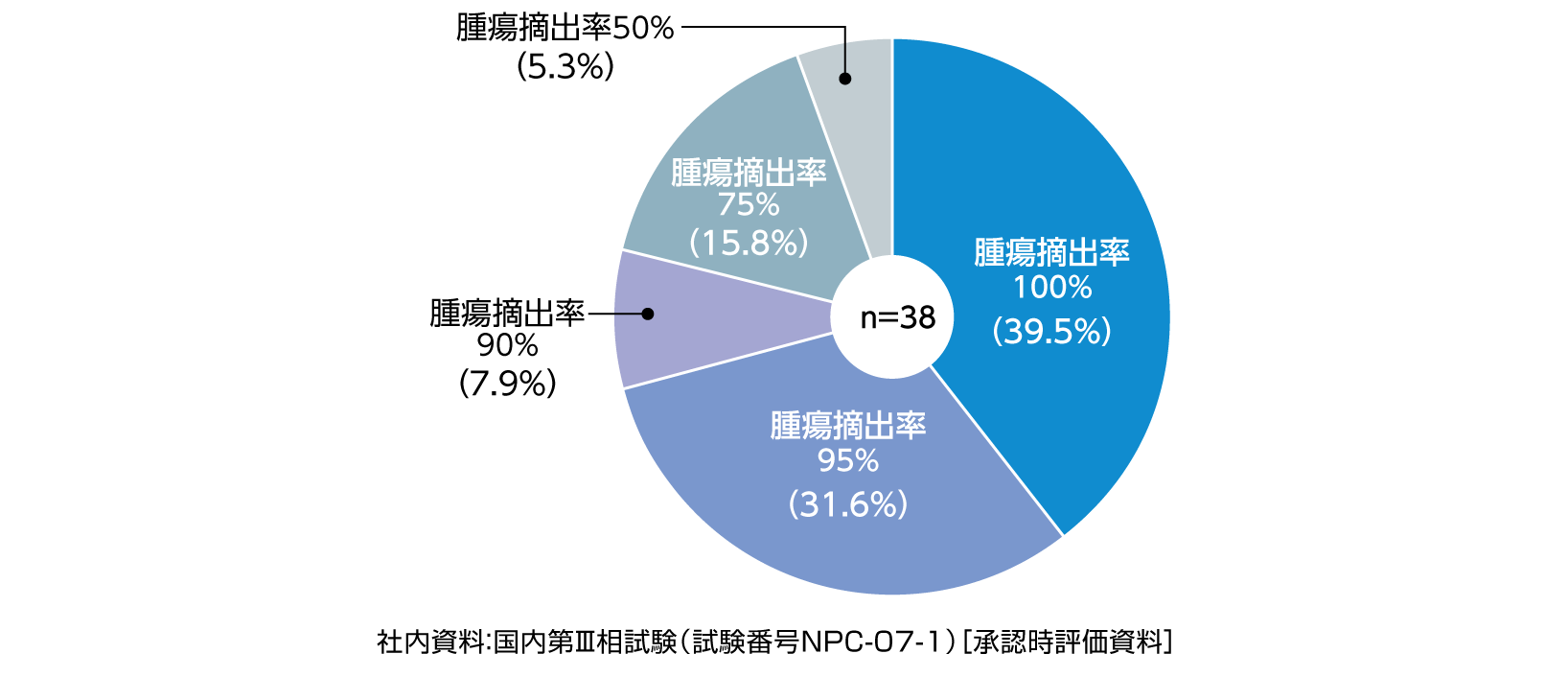
残存腫瘍のない患者の割合(副次評価項目)
術後72時間以内にMRI検査を実施し、腫瘍の摘出率を患者ごとに判定した結果は右図のとおりで、残存腫瘍がない腫瘍摘出率100%の患者は39.5%
(15/38例、95% CI: 24.0〜56.6%)でした。腫瘍摘出率の分布から、腫瘍摘出率が95%以上の患者は71.1%で、腫瘍摘出率が50%未満の患者は認められませんでした。
◆
蛍光近接領域(非蛍光)及び腫瘍からの遠隔領域(非蛍光)における
生検組織ごとの陽性診断率(副次評価項目)
蛍光領域に近接した非蛍光領域及び腫瘍から遠隔の非蛍光領域における生検組織ごとの陽性診断率を検討しました。
生検組織ごとの陽性診断率は、近接領域では61.1%(95% CI: 48.9〜72.4%)、遠隔領域では47.5%(95% CI: 34.6〜60.7%)でした。非蛍光である近接領域及び遠隔領域のいずれの領域においても、腫瘍細胞が浸潤していることが認められ、非蛍光領域に多数の腫瘍細胞が浸潤していることが示されました。

(4)
安全性
安全性評価対象集団の45例における副作用(臨床検査値異常を含む)発現例数は11例(24.4%)で、悪心3例(6.7%)、嘔吐2例(4.4%)、発熱2例(4.4%)、肝機能異常2例(4.4%)、LDH増加1例(2.2%)、γ-GTP増加1例(2.2%)、リンパ球数減少1例(2.2%)、血小板数減少1例(2.2%)、血尿1例(2.2%)でした。重篤な副作用は、肝機能異常、血小板数減少、発熱が各1例に発現しました。
このうち、肝機能異常を発現した1例が死亡に至りましたが、当該死亡事象とは、治験担当医により投与薬剤との因果関係なしと判断されました。
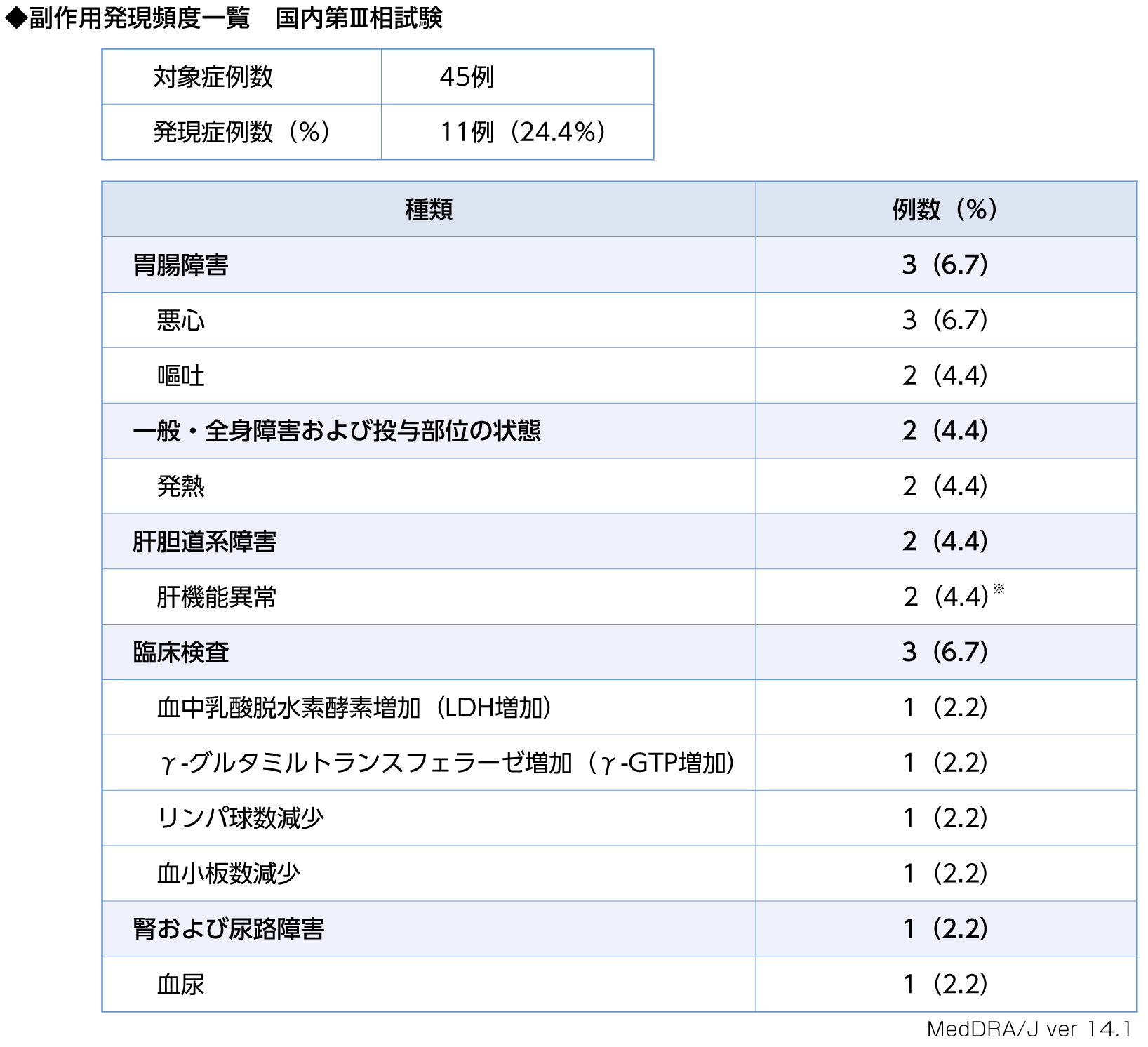
| ※ |
肝機能異常例で、異常変動と判定された2例の臨床検査項目の内訳
1例:γ-GTP増加、AST増加、ALT増加、AL-P増加
1例:γ-GTP増加、AST増加、ALT増加
|
- 社内資料:国内第Ⅲ相試験(試験番号NPC-07-1)[承認時評価資料]

